Common cancer therapy technique like chemotherapy is taking the advantage of apoptosis to eliminate malignant cells within tumors. Epigenetic changes in living cell involve modulations in the expression levels of genes and are recognized as integral to the pathogenesis of many diseases including cancer, depression and various neurogenerative diseases. Reversible modification of histone protein carried out by histone acetyl transferase (HAT) and histone deacetylase (HDAC) enzymes, is involved in the chromatin remodeling and play important role in gene regulation. Over activity of certain HDACs was observed with several cancers, which mediate transcriptional repression by condensing the structure of chromatin and restricting the access of transcriptional factors and thus lead to gene silencing and tumor growth. Inhibition of their activity can induce cell cycle arrest, differentiation and/or apoptosis in cancer cells. Therefore, today HDACs are potential targets and identification of potent HDAC inhibitors possessing adequate ‘drug like’ properties is a considerable effort to develop therapeutics for the treatment of cancer.
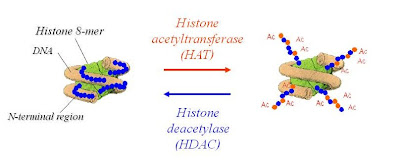 Figure 1. Reversible histone acetylation mediated by HAT and HDAC.
Figure 1. Reversible histone acetylation mediated by HAT and HDAC.
Discovery of trichostatin A (TSA), isolated from Streptomyces hygriscipicus, as potent HDAC inhibitor by Yoshida and co workers (Riken, Japan) (J. Biol. Chem., 1990, 265, 17174–17179) and further X-ray crystallographic report of TSA bound histone deacetylase like protein (HDLP) by Finnin et al (Nature 401, 188-193), opened this new area of design and synthesis of HDAC inhibitors and their SAR study with HDAC paralogs. Several structurally unrelated natural and synthetic compounds have been reported so far as HDAC inhibitors (Figure 2) (J. Med. Chem., 2003, 46, 5097-5116). Major research groups contributing to this area of research are, Prof. Ronald Breslow (Columbia), Prof. Stuart Schreiber (Harvard), Prof. Alan Kozikowski (Illinois), Prof. Naoki Miyata (Nagoya City University, Japan), Dr. Thomas Miller (Merck), Prof. Norikazu Nishino (Kyushu Institute of Technology, Japan) and researcher from Methylgene (Canada). Prof. Nishino's research group is the pioneer in developing cyclic tetrapeptide based HDAC inhibitors. Their research is focused on both modification macrocylic cap group as well as incorporation of many zinc binding ligands to develop many analogs of natural cyclic peptides like chlamydocin, trapoxin and cyl-1 etc.
 Figure 2. Natural and synthetic HDAC inhibitors.
Figure 2. Natural and synthetic HDAC inhibitors.
The suberoylanilide hydroxamic acid (SAHA) also called Vorinostat, is another small molecular non-peptide class of HDAC inhibitor invented by Prof. Ronald Breslow and co workers (Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 5705-5708), which is the first HDAC inhibitor that was been approved by FDA in the year 2006 as an anticancer drug. This molecule is being manufactured and marketed by Merck and Co. with trade name Zolinza. It is used specially for the treatment of T-cell lymphoma (CTCL). In the following video one can see the animation of action of HDAC enzyme and its inhibition by Vorinostat molecule.



Apart from SAHA, many small molecular HDAC inhibitors are in pipeline, undergoing phase clinical trials and among them Panobinostat (LBH-589), developed by Novartis, is in phase clinical trial III. Panabinostat is being observed as potent active molecule against breast and prostate cancers. Hopefully this will be the second HDAC inhibitor coming to the market as anticancer drug in near future. Other HDAC inhibitors which are in clinical trial are Romidepsin, Mocetinostat and Belinostat etc.
Recently, Prof. H. Luesh and co-workers at University of Florida have isolated and characterized the depsipeptide named Largazole from a marine cyanobacterium Symploca sp (JACS, 2008, 130, 1806-1807), consisting of an unusual 16-membered macrocycle incorporated with linearly fused 4-methylthiazoline and thiazole with an ester of a 3-hydroxy-7-mercaptohept-4-enoic acid unit, which constitutes the side chain showing an essential unit for the potent HDAC inhibition (Figure 3).

Figure 3. Structure of Largazole.
Unlike other cyclic tetrapeptide based macrocylic HDAC inhibitors, it was found inhibiting selectively the HDAC-1 paralog. Several research groups including Prof. H. Luesh have also reported the total synthesis of Largazole (JACS, 2008, 130, 8455-8459; JACS, 2008, 130, 11219-11222; Org. Lett. 2008, 10, 3907-3909; Org. Lett. 2010, 12, 1368-1371). This intriguing yet chemically tractable structure responsible for its interesting biological activity warrants further investigation for the therapeutic potential or its next-generation analogs. In recent years some research groups have reported synthesis of largazole analogs and their SAR studies (Org. Lett., 2008, 10, 4021–4024; Org. Lett., 2009, 11, 1301-1304; ChemMedChem. 2009, 4, 1269-1272). However there is still large scope for the development of its analogs and their SAR study to find potent and selective HDAC inhibitors.
Although the precise function of each HDAC paralog is unknown, evidence suggests that they are involved in diverse biological activities, like apoptosis. Therefore, finding selective inhibitor for these HDAC paralogs, either by modifying the cap group region inhibitor or investigating better ligand that can selectively co-ordinate with zinc ion present in the active site of HDAC paralogs, is the basic thrust in this area of research.
No comments:
Post a Comment
Note: Only a member of this blog may post a comment.